Hilfe beim Lesen des Chromatogramms
Pause
In diesem Chromatogramm findet sich eine genetische Variation:

Dort steht, dass die "Referenzsequenz" die oberste Zeile ist und dass ich den allgemeinen genetischen Code verwenden kann, um den Leserahmen zu finden.
Ich kann sehen, dass es zwei N statt einem G und einem C gibt. Ich weiß jedoch nicht, was das bedeutet. Ich bin mir auch nicht sicher, wie ich den Leserahmen finden würde? Ich nehme an, ich könnte nach einem Start- und Stoppcodon suchen, aber ich könnte sie in beide Richtungen lesen?
Letzte Frage: Ist diese Veränderung wahrscheinlich pathogen? Ich bin mir nicht sicher, wie ich eine Variation als pathogen einstufen soll oder nicht.
Antworten (2)
cagliari2005
Die Spuren, die Sie haben, stammen von der Sanger-Sequenzierung . N in der Genetik bedeutet Nukleotid (überraschend, oder?). N wird verwendet, wenn die Base an einem bestimmten Ort unbekannt ist (oder ein beliebiges Basenpaar sein könnte).
In Ihrem Fall haben Sie Ns, weil die Base-Calling-Software das Nukleotid nicht bestimmen kann. Das erste N ist darauf zurückzuführen, dass sich zwei Spitzen überlappen (G- und A-Signale), und das zweite sollte eher ein C als ein N sein.
Soweit ich sehen kann, haben Sie keine offensichtlichen Mutationen, also ist es unwahrscheinlich, dass diese Veränderungen pathogen sind. Eine Mutation kann aufgerufen werden, wenn Sie wissen, dass eine Base in der Referenz in Ihrer Probe modifiziert ist, was hier nicht der Fall ist (einfach N -> G und ein N -> C). Die Signale für Ihre Ns sehen zwischen Ihrer Referenz und Ihrer Probe sehr ähnlich aus und deuten daher nicht auf eine Mutation hin.
Für den Leserahmen müssen Sie nach einem Start- (ATG) und einem Stoppcodon (TAG, TAA oder TGA) suchen, um den offenen Leserahmen (ORF) zu identifizieren. Mehrere Software tun dies und hier ein Link zu einem Online-NCBI-Tool namens ORF Finder .
Wie Sie schon sagten, kann der ORF auch im Gegenstrang sein. Sie möchten nicht die umgekehrte Sequenz betrachten, sondern die umgekehrte komplementäre Sequenz. Normalerweise haben Werkzeuge zur Erkennung von ORFs die Möglichkeit, beide Stränge zu betrachten.
Der Nachtmann
Die beiden Ns, die Sie sehen, sind also nicht unbedingt Varianten, sondern wahrscheinlich nur Lesevorgänge von schlechter Qualität. Wenn Sie DNA sequenzieren und der Sequenzer nicht sagen kann, was die Base ist, wird er sie nur als N bezeichnen, was bedeutet, dass die Base eine der vier DNA-Basen sein könnte.
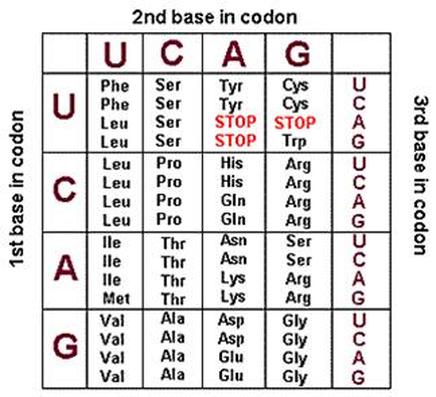
Wenn Sie wissen, dass diese Sequenz ein Stoppcodon enthält, hilft das, um den Messwert zu finden, und suchen Sie einfach nach einer der Stoppsequenzen. Wenn nicht, suchen Sie nach Basen, die mit Codonsequenzen übereinstimmen, und wenn Sie einen vollständigen Sequenzstrang erhalten, der für Aminosäuren codiert, liegen Sie wahrscheinlich richtig.
Als Referenz finden Sie hier einen Link zu einer Codon-Tabelle, die Ihnen bei der Entschlüsselung des Leserahmens hilft.
{kind=link}
Hilft das?
Schreiben Sie die Haplotypen der Familie auf
Stammbaumproblem und Art der Vererbung [geschlossen]
Der Stammbaum der X-rezessiven Störung verwirrt mich
Problem bei der DNA-Sequenzierung
Satz von Bayes für Mutationen
X-rezessive Erkrankungen und genetische Marker
Was ist ein genetischer Marker?
DNA-Mutationen in CHO-KI-Säugerzellen
Frage zu autosomal rezessiven Allelen
Kann der DNA-Test des Bruders meiner Großeltern meine Abstammung von diesem Zweig der Familie offenbaren?
Pause
Der Nachtmann